
最经典、主流的药物靶点筛选技术——TPP热蛋白组分析
阅读:2991
时间:2025-06-03
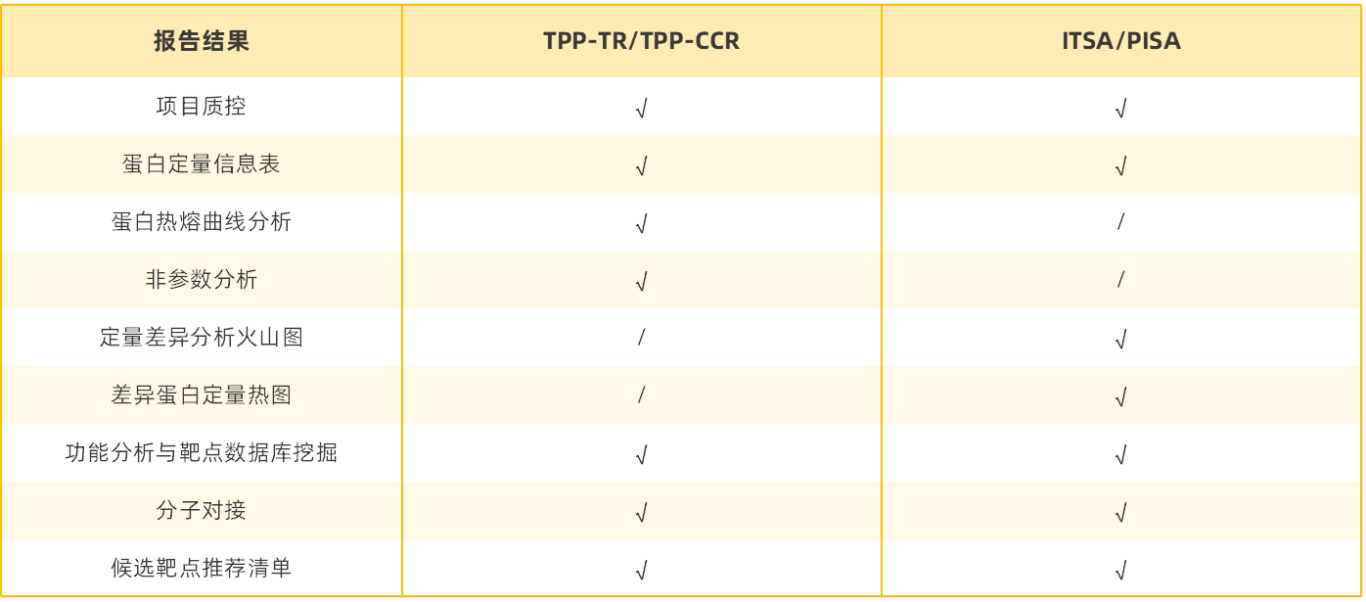
热蛋白组分析技术(Thermal Proteome Profiling,TPP)是一种基于蛋白质热稳定性变化的原理来研究蛋白质-小分子相互作用、蛋白质功能调控及疾病机制的高通量靶点筛选技术。本公司推出的TPP药物靶点筛选解决方案,包含以下几种实验方案:
经典流程TPP-TR(温度梯度蛋白热位移分析)
通过温度梯度(Temperature Range, TPP-TR)实验,构建蛋白热熔曲线,分析药物处理组与对照组间的蛋白热位移(ΔTm),筛选潜在药物作用靶点。
经典流程TPP-CCR(化合物浓度梯度蛋白热位移分析)
过化合物浓度梯度(Compound Concentration Range, TPP-CCR)实验,构建蛋白热熔曲线,分析最高浓度药物处理样本与对照样本间发生显著稳定性变化的蛋白,筛选潜在药物作用靶点。
简化流程TPP-ITSA(等温位移分析)
通过等温位移分析(Isothermal Shift Assay, ITSA)实验,比较药物处理组与对照组在单温度刺激下的蛋白变性水平差异(热稳定性变化),筛选潜在药物作用靶点。
简化流程TPP-PISA(蛋白质组积分溶解度改变)
通过蛋白质组积分溶解度改变(Proteome Integral Solubility Alteration, PISA)实验,比较药物处理组与对照组在多个温度刺激下的混合样本的蛋白变性水平差异(热稳定性变化),筛选潜在药物作用靶点。
蛋白质在加热过程中会逐渐去折叠(变性),暴露出内部疏水氨基酸残基,进而发生沉淀。配体结合会改变蛋白质的热稳定性,导致其熔解温度(Tm,蛋白发生一半变性沉淀时的温度)发生偏移。需要注意的是,大多数蛋白与配体结合后稳定性提高,表现出正向变化,但也有少数蛋白与配体结合后稳定性降低,表现出负向变化。

图1 热位移现象
进一步地,研究者们发现热位移现象也适用于细胞、组织等复杂体系(图2)。将这一原理与高通量蛋白质检测技术相结合,形成了热蛋白组分析技术(Thermal Proteome Profiling,TPP)。
TPP技术利用高通量质谱系统性检测蛋白质热稳定性变化,用于研究蛋白-配体相互作用、蛋白质复合物形成、翻译后修饰(PTMs)等可影响蛋白热稳定性的生理过程。其核心原理是:蛋白质的热稳定性会因其结合配体或发生修饰而改变,质谱技术可以对蛋白组在每个温度点的变性情况进行定量检测,进而得到所有蛋白在药物作用前后的热稳定性变化,揭示蛋白质功能状态。
在TPP实验中,需要设置药物处理组与溶剂处理对照组,对两组样本进行热处理。对热处理之后的非变性组分(离心上清)进行质谱检测,筛选出组间蛋白定量差异,分析差异蛋白的热稳定性变化,最终确认药物作用靶点(图3)。
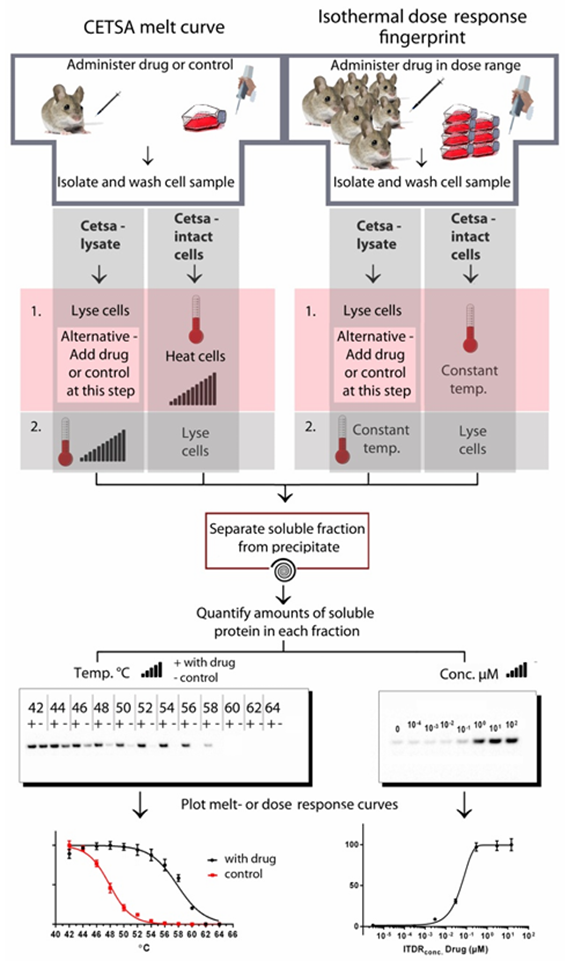
图2 细胞热位移实验

图3 TPP技术流程图
然而,传统的TPP存在一些局限性,如需要大量样本、实验处理流程复杂、质谱检测费用高等。为了避免这些问题,研究者开发了简化流程TPP实验——等温位移分析(ITSA)技术和蛋白质组积分溶解度改变(Proteome Integral Solubility Alteration, PISA)技术。
传统TPP技术通过梯度温度热处理检测熔解温度Tm的变化,而ITSA实验聚焦于特定温度下配体对蛋白质的热稳定性影响。如图4所示,在蛋白发生热位移区域的单个温度条件下,药物处理组与对照组样本的可溶性蛋白水平会存在差异,通过比较单个温度处理条件下的蛋白定量差异即可筛选药物作用靶点。PISA技术利用数学中积分的思想,将多个各温度点的样本进行混合,得到一定温度范围内的药物处理组与溶剂对照组的累计蛋白定量差异,避免了温度点选择的偏差。ITSA和PISA技术能减少成本,简化实验处理流程,适合于高通量筛选。
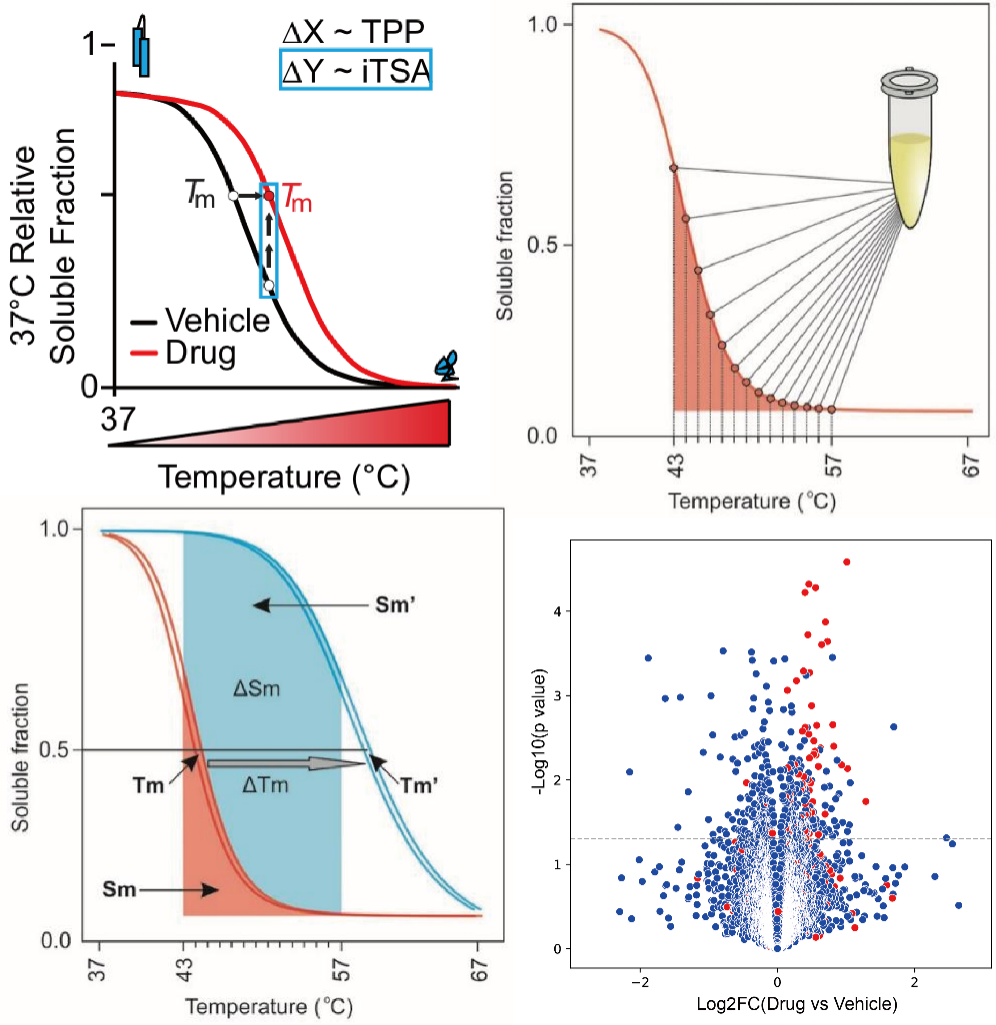
图4 简化流程TPP
影响蛋白热稳定性的因素除了蛋白-配体相互作用之外,还包括蛋白质翻译后修饰、蛋白-蛋白互作、蛋白异构体等(图5)。对于药物活体处理实验,药物作用会引起靶蛋白及其下游蛋白的代谢功能变化,此时通过TPP实验筛选出的热稳定性变化蛋白可能包括药物直接作用靶点、发生翻译后修饰和互作变化的下游靶点等。而在体外药物处理实验中,由于药物直接对蛋白裂解液进行短时间处理,避免了代谢过程,筛选出的热稳定性变化蛋白更多的是直接的药物作用靶点。
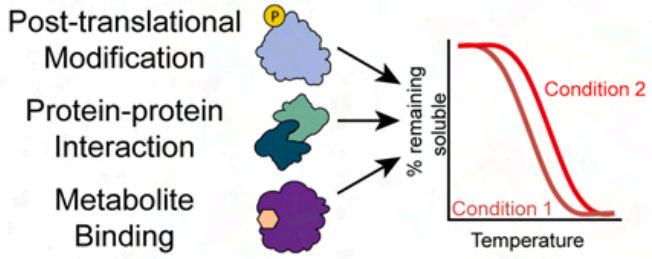
图5 影响蛋白热稳定性变化因素
简单便捷
无需设计合成分子探针;提供细胞/组织和药物分子就可以进行实验。
适用性强
TPP技术适用于多种类型的药物分子(小分子化合物、多肽等)与实验条件(体内外药物处理)。
数据驱动
利用高通量的质谱技术,无偏见地筛选数据,保障获得真实的阳性结果,实现治疗靶点与脱靶靶点的全覆盖。
直接结论
通过比较筛选、生信分析和数据库挖掘,报告直接给出推荐的候选靶点列表清单。
验证数据
对潜在靶蛋白与药物分子进行分子对接,在计算机模拟层面给药物靶点进行验证。
MAIN CONTENT
常规流程TPP
客户提供野生型或疾病模型的细胞/组织及药物(或经药物处理后的细胞/组织),谱度众合实验室在非变性条件下提取样本中的蛋白,随后进行TPP实验。此常规流程TPP需要在37-67℃(哺乳动物蛋白变性范围)间设置6-10个温度处理点,将药物处理组与溶剂处理组样本分别置于这些温度点下进行热处理。通过离心分离各样本中的变性组分(蛋白沉淀),通过质谱对各样本中溶液上清中的蛋白进行定量检测,分析药物作用后蛋白组的热位移变化,筛选药物作用靶点。
1.12-40个样本((药物处理组+溶剂对照组)* Pt(温度梯度点);如需增加不同浓度或者不同药物处理组,每组增加Pt个样本);理论上TPP各实验组在不同温度处理下的样本可以看作重复,因此常规流程TPP通常可以不设置重复实验或者仅设置两组重复。
2.实验组与对照组每组细胞总量建议>2*10^7(组织样本>10mg);药物用量:1μmol左右粉末或20μL药物母液(100×处理浓度)。药物质量计算:1μmol =(摩尔分子质量) μg,例如分子量1000 Da的药物,需要1mg的量。
3.收集细胞/组织样本时需以PBS清洗干净,保留细胞沉淀/去浮水组织,样本不含有任何去垢剂/变性剂成分。
简化流程TPP
客户提供原始的野生型或疾病模型的细胞/组织及药物,谱度众合实验室在非变性条件下提取样本中的蛋白,对蛋白提取物进行药物/溶剂对照处理。对样本进行单温度热处理(哺乳动物样本建议48-56℃),通过离心分离各样本中的变性组分(蛋白沉淀),通过质谱对各样本中溶液上清的蛋白进行定量检测,分析药物作用后蛋白组的热稳定性变化,筛选药物作用靶点。
1.6-10个给药前样本(药物处理组与溶剂对照组各3-5个重复;如需增加不同浓度或者不同药物处理组,每组增加3-5个样本)。
2.实验组与对照组每组细胞总量建议>1*10^7(组织样本>5mg);药物用量:1μmol左右粉末或10μL药物母液(100×处理浓度)。
3.收集细胞/组织样本时需以PBS清洗干净,保留细胞沉淀/去浮水组织,样本不含有任何去垢剂/变性剂成分。
我们将TPP的实验结果分成四个板块进行展示。其中第一部分是质谱检测结果的质控;第二部分TPP数据分析及候选靶点筛选结果,也是文章里使用的主要结果;第三部分对筛选出的候选靶点进行功能富集分析与潜在靶点挖掘,辅助我们进行靶点筛选与验证。第四部分分子对接提供靶蛋白与药物分子的结合能信息,可以作为辅助验证的结果。

图6 项目质控
项目质控结果,包括肽段长度分布、漏切位点分布、蛋白肽段数目分布,肽段电荷分布。符合理论预期的质控结果能证明项目在实验处理和质谱检测阶段的稳定性。
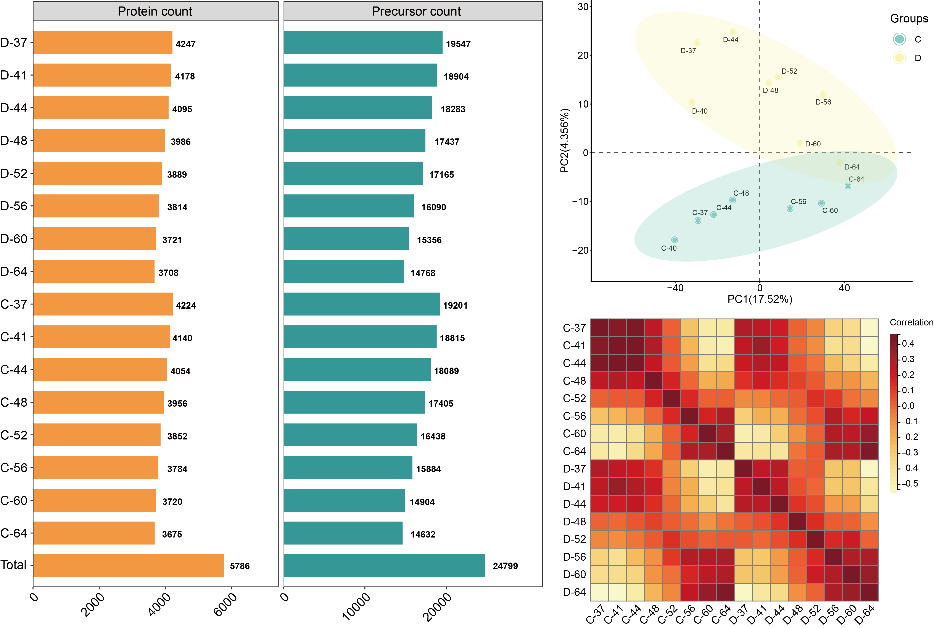
图7 项目基础数据
展示项目定量到的unique肽段数目及其归属的unique蛋白。TPP实验中蛋白鉴定和定量水平会呈现出温度梯度变化。

图8 TPP蛋白热变性曲线分析与非参数分析
对搜库数据进行预处理,以各组37℃的样本蛋白定量值为基准,计算各温度处理下蛋白剩余组分,对预处理后的数据进行分析。包括蛋白热熔曲线分析(通过比较实验组与对照组的ΔTm来筛选靶点)与非参数分析(通过统计学差异分析筛选靶点)两种方式。

图9 ITSA蛋白定量分析
对药物处理组与溶剂对照组样本的蛋白定量数据进行差异倍数计算和T检验,筛选出药物作用后发生热稳定性变化的蛋白,即为潜在的药物作用靶点。
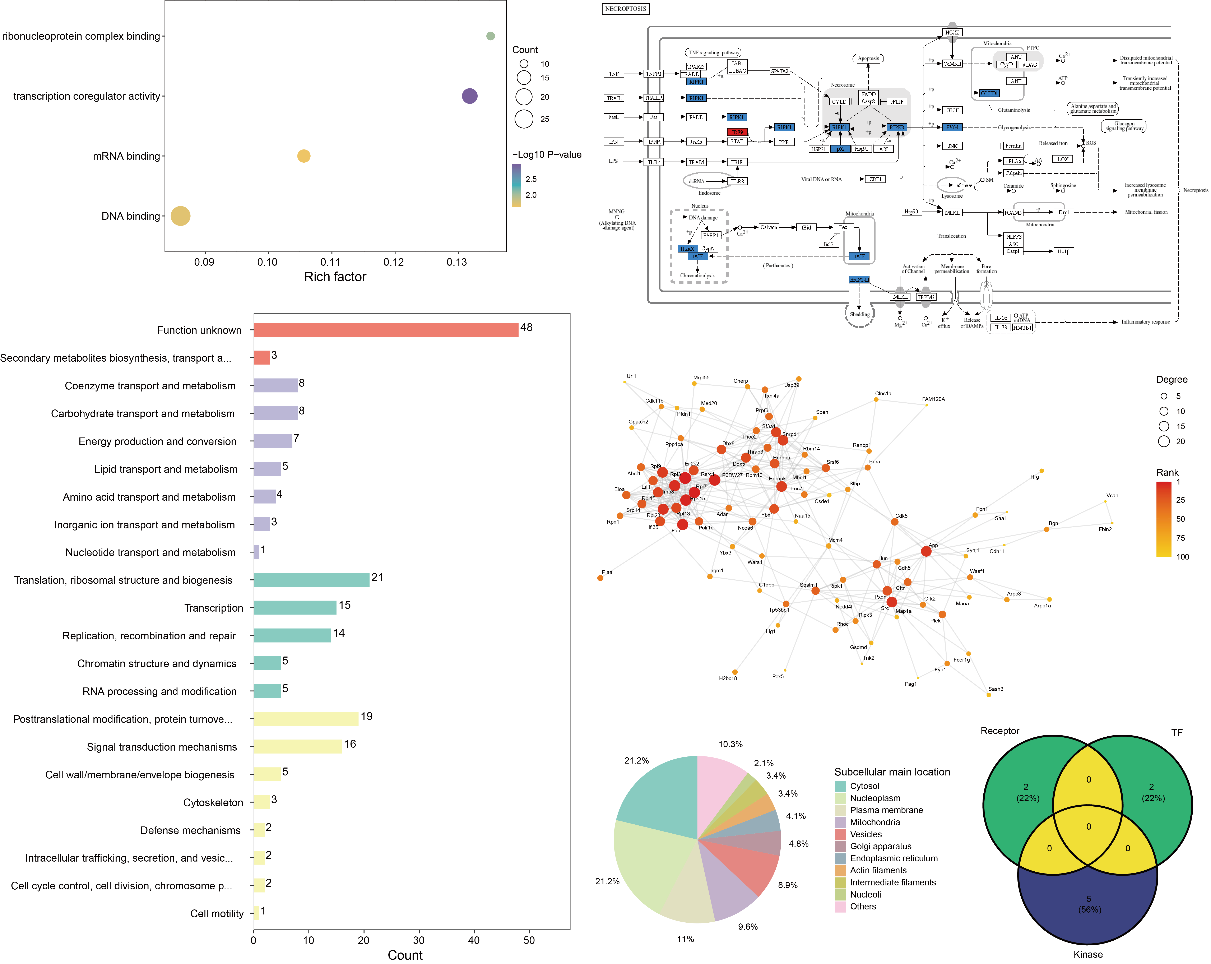
图10 功能分析与潜在靶点挖掘
注释富集分析,项目中包括GO、KEGG富集分析,蛋白互作网络(PPI)分析。潜在靶点挖掘。这些分析内容可以为进一步的靶点筛选与验证提供理论支持。潜在靶点挖掘:从AnimalTFDB4/PhosphoSitePlus/TCDB/TTD/CellChatDB等数据库中对实验所鉴定到的差异蛋白中的转录因子、激酶、离子通道蛋白、药物靶点和受体蛋白等潜在药物靶点蛋白进行注释。
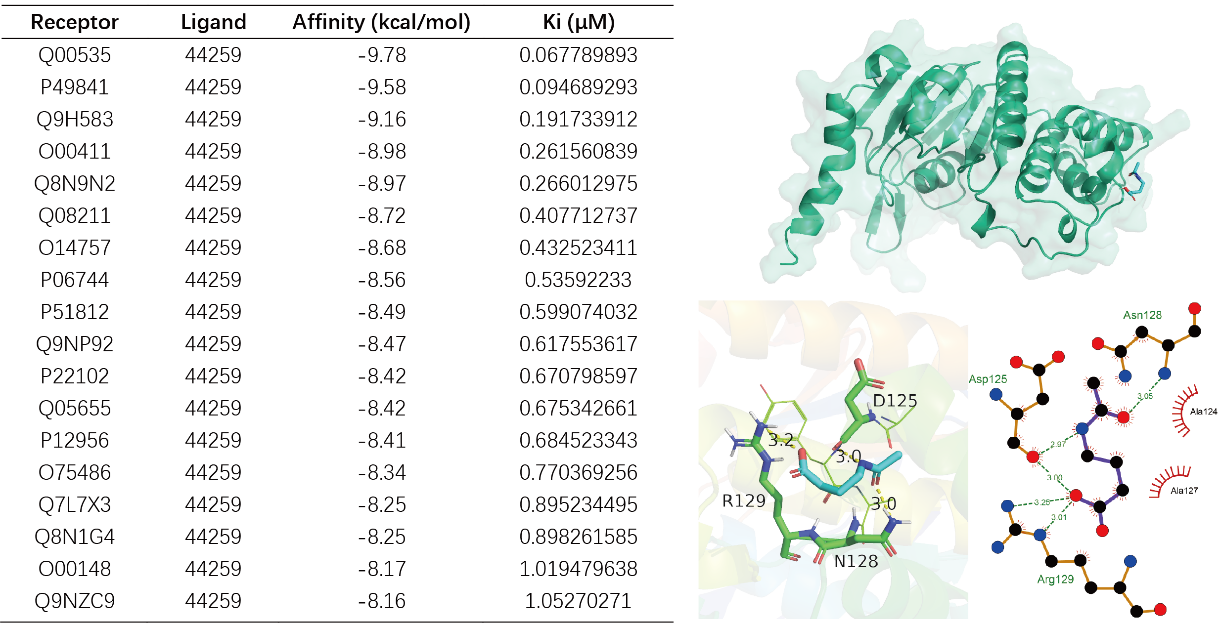
分子对接通过计算机模拟计算药物和蛋白的结合能及结合位点,能为药物和蛋白的相互作用补充证据。
总结
谱度众合是一家由武汉大学博士团队创办的科研服务企业,我们专注于利用质谱技术服务于生物标志物、药物靶点筛选、基础研究等蛋白质研究领域,我们在本专业细分领域持续深耕十几年,针对具体研究场景开发多种面向具体研究目的和论文发表需求的服务产品,助力于客户更轻松更高效的完成科研目标。
撰稿人:程锐锋
审核人:刘宜子、韩强强、肖宇琴
参考文献
[1]. Matulis D, et al. Biochemistry 44(13), 5258-66 (2005).
[2]. Fedorov O, et al. Proc Natl Acad Sci U S A 104(51), 20523-8 (2007).[3]. Daniel Martinez Molina et al. Science 341, 84-87 (2013).
[4]. Mikhail M. Savitski et al..Science 346, 1255784 (2014).
[5]. Franken, H et al. Nat Protoc 10, 1567–1593 (2015).
[6]. Ball, K.A. et al. Commun Biol 3, 75 (2020).
[7]. Gaetani M, et al. J Proteome Res. 18(11), 4027-4037 (2019).
[8]. Amy L. George et al. J Proteome Res. 22 (8), 2629-2640 (2023).[9]. Childs, Dorothee et al. Molecular & cellular proteomics 18(12), 2506-2515 (2019).

















